Cell Arrays¶
Cell Arrays is a Python package that provides a TileDB-backed store for large collections of genomic experimental data, such as millions of cells across multiple single-cell experiment objects.
Install the package¶
To get started, install the package from PyPI
pip install cellarr
Build the CellArrDataset¶
The CellArrDataset method is designed to store single-cell RNA-seq
datasets but can be generalized to store any 2-dimensional experimental data.
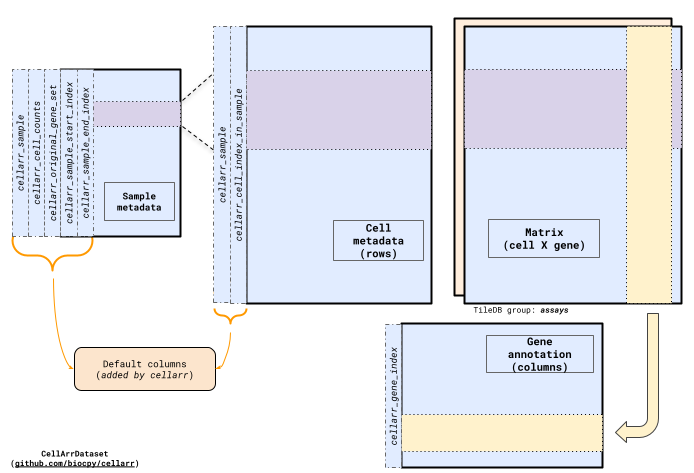
This method creates four TileDB files in the directory specified by output_path:
gene_annotation: A TileDB file containing feature/gene annotations.sample_metadata: A TileDB file containing sample metadata.cell_metadata: A TileDB file containing cell metadata including mapping to the samples they are tagged with insample_metadata.A matrix TileDB file named by the
layer_matrix_nameparameter. This allows the package to store multiple different matrices, e.g. normalized, scaled for the same cell, gene, sample metadata attributes.
The TileDB matrix file is stored in a cell X gene orientation. This orientation
is chosen because the fastest-changing dimension as new files are added to the
collection is usually the cells rather than genes.
Process:
Scan the Collection: Scan the entire collection of files to create a unique set of feature ids (e.g. gene symbols). Store this set as the
gene_annotationTileDB file.Sample Metadata: Store sample metadata in
sample_metadataTileDB file. Each file is typically considered a sample, and an automatic mapping is created between files and samples.Store Cell Metadata: Store cell metadata in the
cell_metadataTileDB file.Remap and Orient Data: For each dataset in the collection, remap and orient the feature dimension using the feature set from Step 1. This step ensures consistency in gene measurement and order, even if some genes are unmeasured or ordered differently in the original experiments.
Note
Check out the reference documentation for modifying the parameters for any of these steps.
First lets mock a few AnnData objects:
import anndata
import numpy as np
import pandas as pd
def generate_adata(n, d, k):
np.random.seed(1)
z = np.random.normal(loc=np.arange(k), scale=np.arange(k) * 2, size=(n, k))
w = np.random.normal(size=(d, k))
y = np.dot(z, w.T)
gene_index = [f"gene_{i+1}" for i in range(d)]
var_df = pd.DataFrame({"names": gene_index}, index=gene_index)
obs_df = pd.DataFrame({"cells": [f"cell1_{j+1}" for j in range(n)]})
adata = anndata.AnnData(layers={"counts": y}, var=var_df, obs=obs_df)
return adata
adata1 = generate_adata(1000, 100, 10)
adata2 = generate_adata(100, 1000, 100)
print("datasets")
print(adata1, adata2)
datasets
AnnData object with n_obs × n_vars = 1000 × 100
obs: 'cells'
var: 'names'
layers: 'counts' AnnData object with n_obs × n_vars = 100 × 1000
obs: 'cells'
var: 'names'
layers: 'counts'
/home/runner/work/cellarr/cellarr/.tox/docs/lib/python3.11/site-packages/anndata/_core/aligned_df.py:68: ImplicitModificationWarning: Transforming to str index.
warnings.warn("Transforming to str index.", ImplicitModificationWarning)
/home/runner/work/cellarr/cellarr/.tox/docs/lib/python3.11/site-packages/anndata/_core/aligned_df.py:68: ImplicitModificationWarning: Transforming to str index.
warnings.warn("Transforming to str index.", ImplicitModificationWarning)
To build a CellArrDataset from a collection of H5AD or AnnData objects:
import anndata
import numpy as np
import tempfile
from cellarr import build_cellarrdataset, CellArrDataset, MatrixOptions
# Create a temporary directory
tempdir = tempfile.mkdtemp()
# # Read AnnData objects
# adata1 = anndata.read_h5ad("path/to/object1.h5ad", "r")
# # or just provide the path
# adata2 = "path/to/object2.h5ad"
# Build CellArrDataset
dataset = build_cellarrdataset(
output_path=tempdir,
files=[adata1, adata2],
matrix_options=MatrixOptions(dtype=np.float32),
)
print(dataset)
Optimizing /tmp/tmp7tta1opt/gene_annotation
Fragments before consolidation: 1
Fragments after consolidation: 1
Optimizing /tmp/tmp7tta1opt/sample_metadata
Fragments before consolidation: 1
Fragments after consolidation: 1
Optimizing /tmp/tmp7tta1opt/cell_metadata
Fragments before consolidation: 1
Fragments after consolidation: 1
/home/runner/work/cellarr/cellarr/.tox/docs/lib/python3.11/site-packages/cellarr/build_cellarrdataset.py:247: UserWarning: Scanning all files for feature ids (e.g. gene symbols) and cell annotations, this may take long, Please also make sure you have enough memory.
warnings.warn(
/home/runner/work/cellarr/cellarr/.tox/docs/lib/python3.11/site-packages/cellarr/build_cellarrdataset.py:299: UserWarning: Scanning all files to compute cell counts, this may take long
warnings.warn(
/home/runner/work/cellarr/cellarr/.tox/docs/lib/python3.11/site-packages/cellarr/build_cellarrdataset.py:306: UserWarning: Sample metadata is not provided, each dataset in 'files' is considered a sample
warnings.warn(
/home/runner/work/cellarr/cellarr/.tox/docs/lib/python3.11/site-packages/cellarr/build_cellarrdataset.py:337: UserWarning: Scanning all files for feature ids (e.g. gene symbols), this may take long
warnings.warn(
/home/runner/work/cellarr/cellarr/.tox/docs/lib/python3.11/site-packages/cellarr/build_cellarrdataset.py:358: UserWarning: Scanning all files to compute cell counts, this may take long
warnings.warn(
Optimizing /tmp/tmp7tta1opt/assays/counts
Fragments before consolidation: 2
Fragments after consolidation: 1
class: CellArrDataset
number_of_rows: 1100
number_of_columns: 1000
path: '/tmp/tmp7tta1opt'
Important
All files are expected to be consistent and any modifications to make them consistent is outside the scope of this function and package.
There’s a few assumptions this process makes:
If object in
filesis anAnnDataor H5AD object, these must contain an assay matrix in the layers slot of the object named aslayer_matrix_nameparameter.Feature information must contain a column defined by the parameter
feature_columnin theGeneAnnotationOptions.that contains feature ids or gene symbols across all files.If no
cell_metadatais provided, we scan to count the number of cells and create a simple range index.Each file is considered a sample and a mapping between cells and samples is automatically created. Hence the sample information provided must match the number of input files.
Optionally provide cell metadata columns¶
If the cell metadata is inconsistent across datasets, you can provide a list of
columns to standardize during extraction. Any missing columns will be filled with
the default value 'NA', and their data type should be specified as 'ascii' in
CellMetadataOptions. For example, this build process will create a TileDB store
for cell metadata containing the columns 'cellids' and 'tissue'. If any dataset
lacks one of these columns, the missing values will be automatically filled with 'NA'.
dataset = build_cellarrdataset(
output_path=tempdir,
files=[adata1, adata2],
matrix_options=MatrixOptions(dtype=np.float32),
cell_metadata_options=CellMetadataOptions(
column_types={"cellids": "ascii", "tissue": "ascii"}
),
)
print(dataset)
Query a CellArrDataset¶
Users have the option to reuse the dataset object retuned when building the dataset or by creating a CellArrDataset object by initializing it to the path where the files were created.
# Create a CellArrDataset object from the existing dataset
dataset = CellArrDataset(dataset_path=tempdir)
# Query data from the dataset
expression_data = dataset[10, ["gene_1", "gene_10", "gene_500"]]
print("matrix slice:")
print(expression_data.matrix)
print("\n\n gene_annotation slice:")
print(expression_data.gene_annotation)
print("\n\n cell_metadata slice:")
print(expression_data.cell_metadata)
matrix slice:
{'counts': <COOrdinate sparse matrix of dtype 'float32'
with 2 stored elements and shape (1, 3)>}
gene_annotation slice:
cellarr_gene_index
0 gene_1
1 gene_10
447 gene_500
cell_metadata slice:
cellarr_sample cellarr_cell_index_in_sample
10 sample_1 10
This returns a CellArrDatasetSlice object that contains the matrix and metadata DataFrame’s along the cell and gene axes.
Users can easily convert these to analysis-ready representations
print("as anndata:")
print(expression_data.to_anndata())
print("\n\n as summarizedexperiment:")
print(expression_data.to_summarizedexperiment())
as anndata:
AnnData object with n_obs × n_vars = 1 × 3
obs: 'cellarr_sample', 'cellarr_cell_index_in_sample'
var: 'cellarr_gene_index'
layers: 'counts'
as summarizedexperiment:
class: SummarizedExperiment
dimensions: (3, 1)
assays(1): ['counts']
row_data columns(1): ['cellarr_gene_index']
row_names(3): ['0', '1', '447']
column_data columns(2): ['cellarr_sample', 'cellarr_cell_index_in_sample']
column_names(1): ['10']
metadata(0):
/home/runner/work/cellarr/cellarr/.tox/docs/lib/python3.11/site-packages/anndata/_core/aligned_df.py:68: ImplicitModificationWarning: Transforming to str index.
warnings.warn("Transforming to str index.", ImplicitModificationWarning)
/home/runner/work/cellarr/cellarr/.tox/docs/lib/python3.11/site-packages/anndata/_core/aligned_df.py:68: ImplicitModificationWarning: Transforming to str index.
warnings.warn("Transforming to str index.", ImplicitModificationWarning)
A single cell dataloader¶
A basic single cell dataloader can be instantiated by using the DataModule class.
from cellarr.dataloader import DataModule
datamodule = DataModule(
dataset_path="/path/to/cellar/dir",
cell_metadata_uri="cell_metadata",
gene_annotation_uri="gene_annotation",
matrix_uri="counts",
label_column_name="label",
study_column_name="study",
batch_size=1000,
lognorm=True,
target_sum=1e4,
)
Users can optionally set a list of studies to be used as validation. If not provided, all studies are used for training. Additionally users may also provide the gene space to train their models.
val_studies = ["study1", "study100"]
gene_list = [
"GPNMB", "TREM2", "LPL", "HLA-DQA1", "CD109",
"IL6ST", "SDC2", "MSR1", "ALCAM", "SLC1A3",
"CD9", "CD59", "MRC1", "SLC11A1", "CPM",
"GPR183", "ITGAX", "HLA-DMB", "NRP2", "SV2C",
"PTPRJ", "EMP1", "HLA-DQB1", "MERTK", "CD52",
"CXCL16", "ABCA1", "HLA-DPB1", "OLR1", "CD83"
]
datamodule = DataModule(
dataset_path="/path/to/cellar/dir",
cell_metadata_uri="cell_metadata",
gene_annotation_uri="gene_annotation",
matrix_uri="counts",
val_studies=val_studies,
label_column_name="label",
study_column_name="study",
gene_order=gene_list,
batch_size=1000,
lognorm=True,
target_sum=1e4,
)
Users can access training cells by index.
datamodule.train_dataset[100]
Batches can be created and examined.
dataloader = datamodule.train_dataloader()
batch = next(iter(dataloader))
expression, labels, studies = batch
The dataloader can then be used in training models. The package provides a simple autoencoder to serve as a template for the user’s own models.
import pytorch_lightning as pl
from cellarr.autoencoder import AutoEncoder
autoencoder = AutoEncoder(
n_genes=len(datamodule.gene_indices),
latent_dim=128,
hidden_dim=[1024, 1024, 1024],
dropout=0.5,
input_dropout=0.4,
residual=False,
)
model_path = "/path/to/model/mymodel/"
params = {
"max_epochs": 500,
"logger": True,
"log_every_n_steps": 1,
"limit_train_batches": 100, # to specify number of batches per epoch
}
trainer = pl.Trainer(**params)
trainer.fit(autoencoder, datamodule=datamodule)
autoencoder.save_all(model_path=model_path)
Check out the documentation for more details.